Pipeline
QurAlis is breaking through the barriers of science in our quest to bring much-needed precision therapies to patients.

QurAlis is the leader in development of precision therapies for amyotrophic lateral sclerosis (ALS). In addition to ALS, QurAlis is advancing a robust precision medicine pipeline to bring effective disease-modifying therapeutics to patients suffering from severe diseases defined by genetics and clinical biomarkers. We are focused on patients who have losses of STATHMIN-2 (STMN2), Kv7, or UNC13A. The QurAlis team is leveraging insights, platforms, and successes in ALS to collaborate and expand our pipeline to other neurodegenerative and neurological diseases, such as epilepsy, pain, frontotemporal dementia (FTD), Fragile X syndrome, and progressive supranuclear palsy (PSP).
Our lead therapeutic product candidates are:
- QRL-201 – a first-in-class molecule for the treatment of ALS that aims to restore STMN2 expression in ALS patients.
- QRL-101 – a potentially best-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS, as well as the treatment of epilepsy and pain.


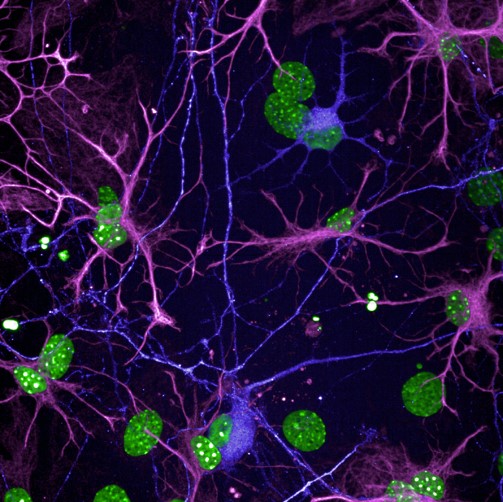
STATHMIN-2
(STMN2) is a well-known and important member of a family of proteins that play critical roles in neural repair, and axon and microtubule stability. The expression of this protein is significantly decreased in ALS patients, contributing to motor neuron loss and cytoskeletal weakness. In animal models, STMN2 deletion was found to cause axonal degeneration, which is the leading functional deficit that drives paralysis in ALS patients.
Kv7.2/7.3
is a voltage-gated potassium channel whose role is crucial for the regulation of neuronal excitability and membrane potential. Kv7.2/7.3 deactivation leads to abnormal electrical activity in the brain. The activation of this channel shows the potential to decrease spinal and cortical/motor neuron excitability and to positively affect CMAP (compound muscle action potential). This suggests that this may be an effective therapeutic approach for ALS patients suffering from hyperexcitability-induced motor neuron degeneration.
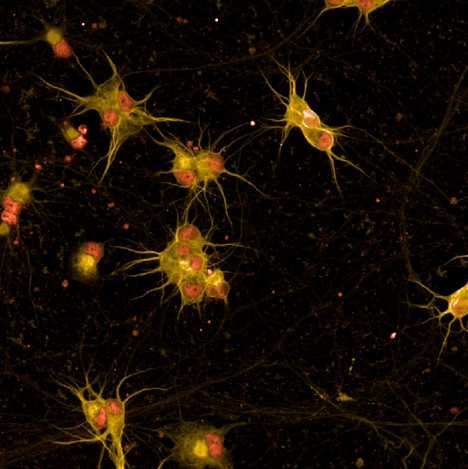
UNC13A
is an essential regulator of neurotransmitter release at synapses. In ALS and FTD, the loss of TDP-43 causes the mis-splicing of certain pre-mRNA transcripts resulting in expression of a cryptic exon-containing transcript that interferes with appropriate protein generation. UNC13A is a pre-mRNA that is mis-spliced due to loss of TDP-43 in disease. Up to 63 percent of ALS patients and up to one-third of FTD patients carry a single nucleotide polymorphism in the UNC13A gene or show TDP-43 pathology which greatly exacerbates UNC13A mis-splicing leading to loss of function of the UNC13A protein.
OUR PLATFORM
FlexASO®: Proprietary Anti-Sense Oligonucleotide Splice Modulator Platform
Bespoke platform key to tackling the spectrum of neurodegenerative and neurological diseases.
Learn More